
FDA Advisory No. 2018-212 || Public Health Warning Against the Purchase and Consumption of the following Unregistered Food Supplements:

ULTIMATE HERBS ACTIVATED CHARCOAL CAPSULE FOOD SUPPLEMENT ULTIMATE HERBS GUYABANO ‘SOURSOP” CAPSULE FOOD SUPPLEMENT ULTIMATE HERBS MANGOSTEEN CAPSULE FOOD SUPPLEMENT ULTIMATE HERBS TURMERIC “LUYANG DILAW” CAPSULE FOOD SUPPLEMENT LIVING NUTRITION […]
FDA Advisory No. 2018-211 || Public Health Warning Against the Purchase and Consumption of the following Unregistered Food Products:

BERYL’S GOURMET MILK COMPOUND NATURAL TURMERIC POWDER ALWAYS FRESH HERRING FILLET MUSTARD SAUCE ALWAYS FRESH HERRING FILLET MANGO PEPPER SAUCE The Food and Drug Administration (FDA) advises the public against […]
FDA Advisory No. 2018-213 || VOLUNTARY RECALL OF STAY SAFE DISINFECTION CAP WITH BATCH NUMBER Z1Z04300

The Food and Drug Administration (FDA) informs the public and all concerned healthcare professionals that Fresenius Medical Care Philippines, Inc. has voluntarily recalled the Stay Safe Disinfection Cap with batch […]
FDA Advisory No. 2018-209 || Dissemination of ASEAN Post-Marketing Alert System (PMAS) Report on Adulterated Cosmetic Products (Reference No. ASEAN ALERT 12-16/2018/K)

The Food and Drug Administration (FDA) hereby issues this public health warning to inform the public of the ASEAN Post-Marketing Alert System (PMAS) report on the following cosmetic products. BRAND […]
CDRR Officers of the Day for 02-04 July 2018

The Center for Drug Regulation and Research (CDRR) will be having the Operational Planning on 02-04 July 218. In line with this, there will be no officers on duty for […]
FDA Advisory No. 2018-206 || VOLUNTARY RECALL OF THE PATHFAST CK-MB AND PATHFAST NTproBNP REAGENTS
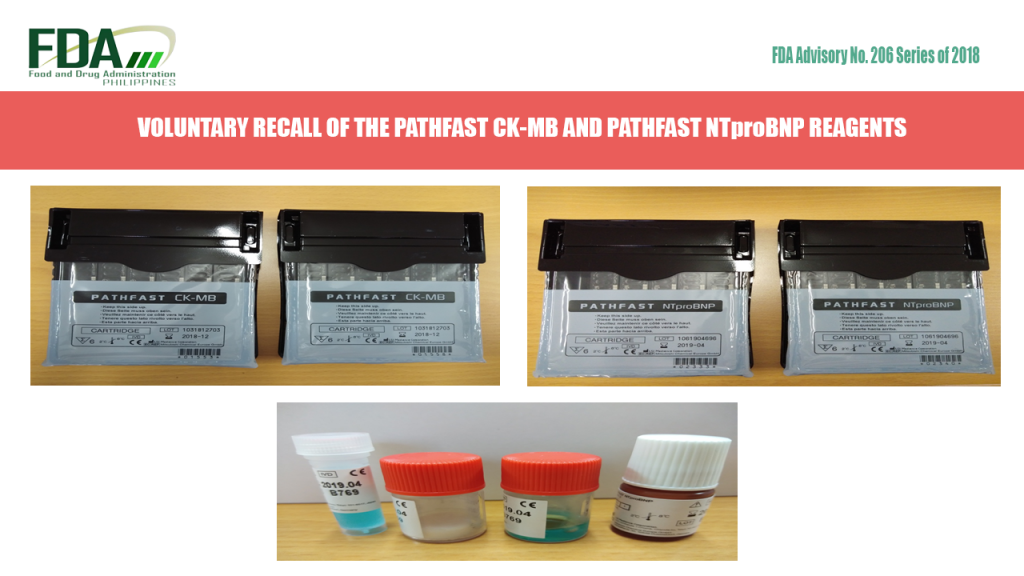
All are hereby advised by the Food and Drug Administration (FDA) on the voluntary recall of PATHFAST CK-MB with LRD/CDRRHR_EXEMP-2015-6374 and PATHFAST NTproBNP with LRD/CDRRHR_EXEMP-2015-6372 distributed by Global Medical Solutions, […]
FDA Advisory No. 2018-207 || VOLUNTARY RECALL OF HARMONIC ACE® LAPAROSCOPIC 5MM DIAMETER SHEARS + ADAPTIVE TISSUE TECHNOLOGY (SPECIFIC LOTS ONLY FOR CODES HAR23 AND HAR36)

The Food and Drug Administration (FDA) informs the public and all concerned healthcare professionals that Johnson & Johnson Philippines, Inc. has voluntarily recalled specific lots of HARMONIC ACE® Laparoscopic 5mm […]
FDA Advisory NO. 2018-205 || Public Health Warning Against the Purchase and Use of Unregistered Medical Devices “Nebulizing Kit” included on the package of Travel Nebulizer of Far East Medical.

The Food and Drug Administration (FDA) advises all concerned healthcare professionals, establishment and general consuming public against the purchase and use of the NEBULIZING KIT included in the package of […]
FDA Advisory No. 2018-208 || Public Health Warning Against the purchase and Use of Unregistered Medical Device Product “Bang-ze Abacterial Flexible Fabric Bandage”

The Food and Drug Administration (FDA) hereby advises the general public and healthcare professionals against the purchase and use of Bang-ze Abacterial Flexible Fabrid bandage. FDA post-marketing surveillance (PMS) activities […]
FDA Advisory No. 2018-197 || Public Health Warning Against the Purchase and Use of Unregistered Medical Device (Bebeta Digital Thermometer)

The Food and Drug Administration (FDA) advises all concerned healthcare professionals and the public against the purchase and use of Bebeta Digital Thermometer: FDA post-marketing surveillance activities have verified that […]